
Localized translation in the heart
 Heart failure is a global problem with an estimated prevalence of 38 million patients worldwide, a number that is increasing with the ageing of the population. Survival after a diagnosis of heart failure has improved during the past 30 years; however, despite the modest improvement, the 5-year mortality is still approximately 50% - worse than that of many cancers. Efforts to enhance the understanding of the pathobiology of heart failure and to develop new approaches for treatments are direly needed. It is often assumed that the structure of the heart is static. We showed that sarcomeres are highly dynamic structures, and even the largest protein, titin, is replaced in vivo across the entire heart with an 8-day half-life.
Heart failure is a global problem with an estimated prevalence of 38 million patients worldwide, a number that is increasing with the ageing of the population. Survival after a diagnosis of heart failure has improved during the past 30 years; however, despite the modest improvement, the 5-year mortality is still approximately 50% - worse than that of many cancers. Efforts to enhance the understanding of the pathobiology of heart failure and to develop new approaches for treatments are direly needed. It is often assumed that the structure of the heart is static. We showed that sarcomeres are highly dynamic structures, and even the largest protein, titin, is replaced in vivo across the entire heart with an 8-day half-life.
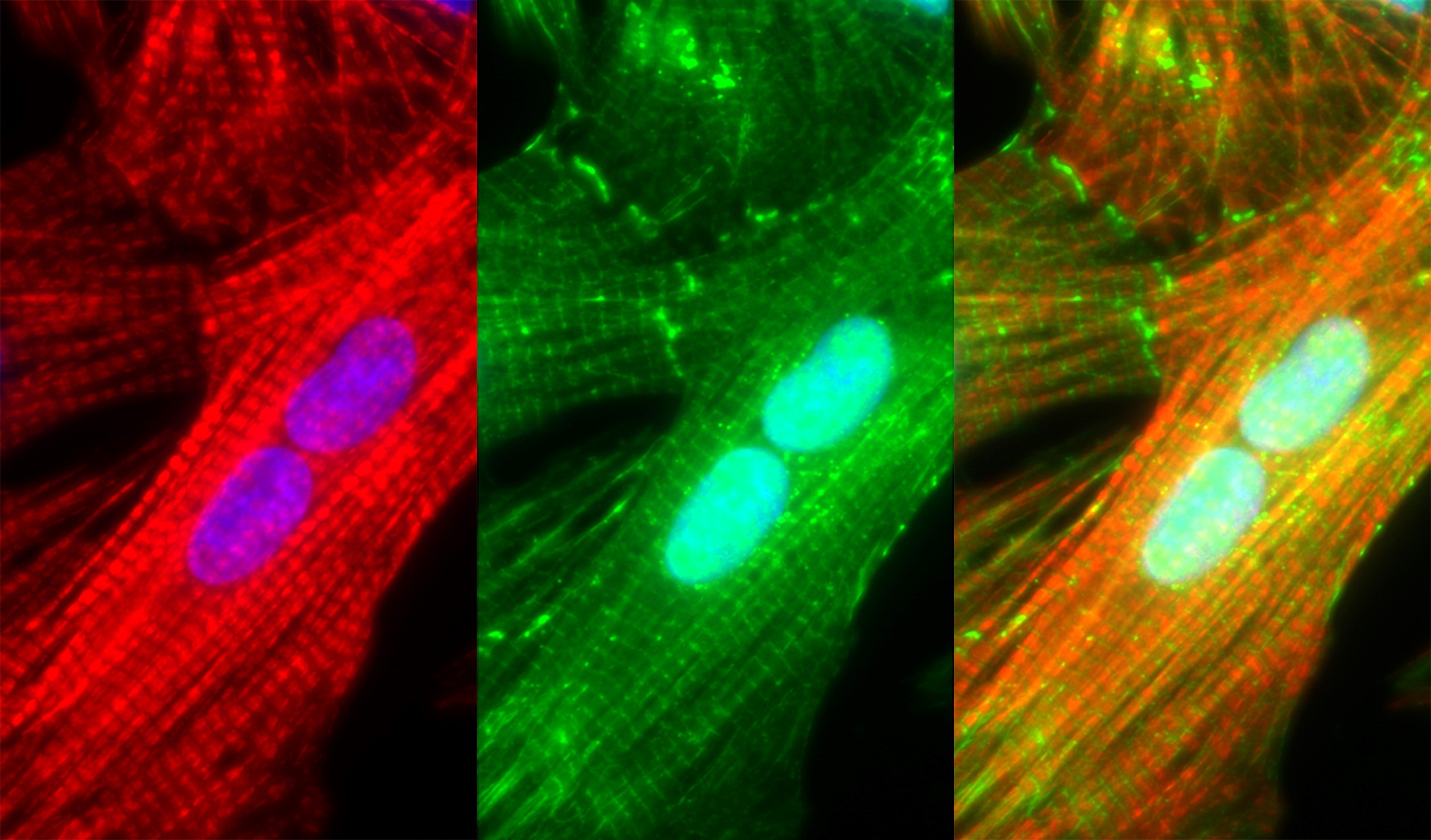 The cells in our bodies often live longer than the proteins they contain, so the proteins need to be constantly replaced. However, it is not entirely clear how large protein complexes, composed of multiple different proteins, maintain their structural and functional integrity while replacing their subunits. This is particularly true for cardiomyocytes, which are long-lived cells whose role is to maintain and operate one major protein complex called the sarcomere. As the human heart is estimated to contain more than three trillion sarcomeric units, maintaining sarcomeres must be an energy-intensive process that has to be performed at a very high fidelity. Indeed, studies in human failing hearts using electron microscopy showed significantly reduced sarcomeric myofibrillar density in patients suffering from either idiopathic or ischemic dilated cardiomyopathies, and proteomic studies confirmed the specific loss of sarcomeric proteins in such failing hearts, suggesting proteostasis failure may play an important role in the progression of heart failure.
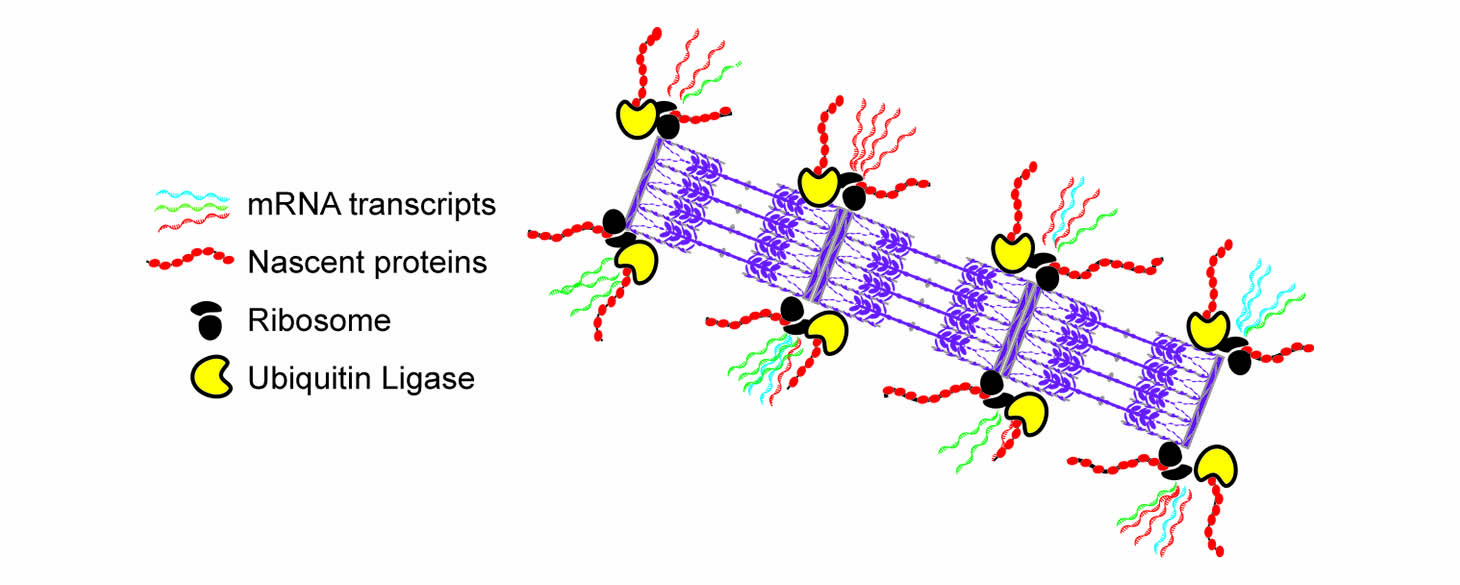
We discovered that sarcomeres contain localized ‘nano-facilities’ positioned on both sides of the Z-line of the sarcomere, where mRNAs, ribosomes, and protein translation are localized. We later identified a ribosomal associated protein called RPSA, and showed that if we knock it out in cardiomyocytes, the cells lose their ribosomal localization, their localized translation, and most of the sarcomeres, showing that localized translation was indeed essential for the maintenance of the sarcomeres. Employing a different labeling scheme of Halo-tagged sarcomeric proteins we imaged and identified the mechanisms underlying the turnover of sarcomeric proteins.
The cells in our bodies often live longer than the proteins they contain, so the proteins need to be constantly replaced. However, it is not entirely clear how large protein complexes, composed of multiple different proteins, maintain their structural and functional integrity while replacing their subunits. This is particularly true for cardiomyocytes, which are long-lived cells whose role is to maintain and operate one major protein complex called the sarcomere. As the human heart is estimated to contain more than three trillion sarcomeric units, maintaining sarcomeres must be an energy-intensive process that has to be performed at a very high fidelity. Indeed, studies in human failing hearts using electron microscopy showed significantly reduced sarcomeric myofibrillar density in patients suffering from either idiopathic or ischemic dilated cardiomyopathies, and proteomic studies confirmed the specific loss of sarcomeric proteins in such failing hearts, suggesting proteostasis failure may play an important role in the progression of heart failure.
We discovered that sarcomeres contain localized ‘nano-facilities’ positioned on both sides of the Z-line of the sarcomere, where mRNAs, ribosomes, and protein translation are localized. We later identified a ribosomal associated protein called RPSA, and showed that if we knock it out in cardiomyocytes, the cells lose their ribosomal localization, their localized translation, and most of the sarcomeres, showing that localized translation was indeed essential for the maintenance of the sarcomeres. Employing a different labeling scheme of Halo-tagged sarcomeric proteins we imaged and identified the mechanisms underlying the turnover of sarcomeric proteins.
Papers:
Lewis YE, Moskovitz A, Mutlak M, Heineke J, Caspi LH, Kehat I. Localization of transcripts, translation, and degradation for spatiotemporal sarcomere maintenance. J Mol Cell Cardiol. 2018 Mar;116:16-28. https://www.ncbi.nlm.nih.gov/pubmed/29371135Scarborough EA, Uchida K, Vogel M, Erlitzki N, Iyer M, Phyo SA, Bogush A, Kehat I, Prosser BL. Microtubules orchestrate local translation to enable cardiac growth. Nat Commun. 2021 Mar 11;12(1):1547 https://pubmed.ncbi.nlm.nih.gov/33707436/
Localized translation and sarcomere maintenance requires ribosomal protein SA in mice. Haddad R, Sadeh O, Ziv T, Erlich I, Haimovich-Caspi L, Shemesh A, van der Velden J, Kehat I. J Clin Invest. 2024 May 14;134(13):e174527 https://pubmed.ncbi.nlm.nih.gov/38743494/
Keeping it fresh: ribosomal protein SA sustains sarcomeric function via localized translation https://pubmed.ncbi.nlm.nih.gov/38949021/
Imaging of Existing and Newly Translated Proteins Elucidates Mechanisms of Sarcomere Turnover. Douvdevany G, Erlich I, Haimovich-Caspi L, Mashiah T, Prondzynski M, Pricolo MR, Alegre-Cebollada J, Linke WA, Carrier L, Kehat I. Circ Res. 2024 Jul 4. https://pubmed.ncbi.nlm.nih.gov/38962864/
Cardiac sarcomere turnover by unidirectional replacement of proteins https://www.nature.com/articles/s41569-024-01065-3
Signaling in heart failure
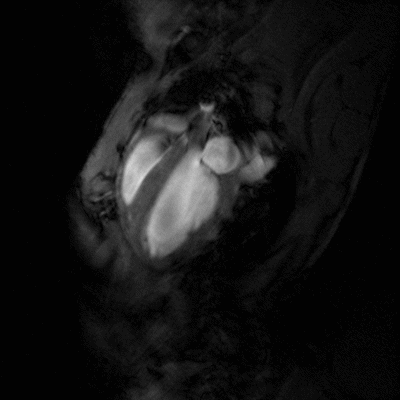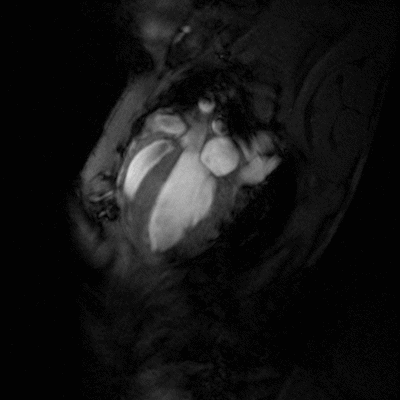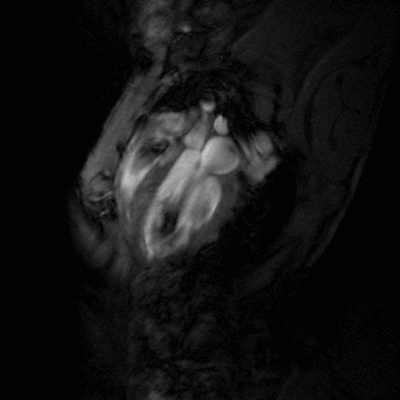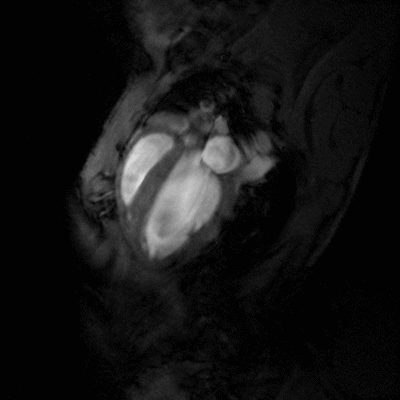 Congestive heart failure (CHF) is a worldwide epidemic. It is estimated, for example, that in Europe around 10 million people are suffering from this disease. Despite some progress in medical treatment within the last 10 years, morbidity and mortality of CHF are still high: 70-80% of patients suffering from heart failure will die within the next 8 years. Heart failure develops mainly after myocardial infarction, chronic arterial hypertension, diseases of the cardiac valves (e.g. aortic valve stenosis), viral myocarditis or genetic disease. Associated with all these different disease entities is a profound alteration of the ventricular shape and function, which is triggered by cardiac overload as well as local and systemic activation of specific cytokines and growth factors. As a final common pathway of almost every heart disease leading to heart failure, the left cardiac ventricle dramatically dilates, while the left ventricular walls become increasingly thinner. This progressive dilation markedly increases wall stress, which in turn leads to further damage to the myocardium and diminishes the capability of the heart to pump blood into the circulation.
Our lab study the molecular mechanisms responsible for cardiac hypertrophy and remodeling during heart failure. We showed that the Extracellular Signal-Regulated Kinase (ERK) pathway acts to promote a compensated hypertrophic response, with enhanced contractile function and reduced fibrosis. The activation of this pathway may be a therapeutic strategy in heart failure to preserve contractile function when the pressure overload cannot be easily alleviated.
Congestive heart failure (CHF) is a worldwide epidemic. It is estimated, for example, that in Europe around 10 million people are suffering from this disease. Despite some progress in medical treatment within the last 10 years, morbidity and mortality of CHF are still high: 70-80% of patients suffering from heart failure will die within the next 8 years. Heart failure develops mainly after myocardial infarction, chronic arterial hypertension, diseases of the cardiac valves (e.g. aortic valve stenosis), viral myocarditis or genetic disease. Associated with all these different disease entities is a profound alteration of the ventricular shape and function, which is triggered by cardiac overload as well as local and systemic activation of specific cytokines and growth factors. As a final common pathway of almost every heart disease leading to heart failure, the left cardiac ventricle dramatically dilates, while the left ventricular walls become increasingly thinner. This progressive dilation markedly increases wall stress, which in turn leads to further damage to the myocardium and diminishes the capability of the heart to pump blood into the circulation.
Our lab study the molecular mechanisms responsible for cardiac hypertrophy and remodeling during heart failure. We showed that the Extracellular Signal-Regulated Kinase (ERK) pathway acts to promote a compensated hypertrophic response, with enhanced contractile function and reduced fibrosis. The activation of this pathway may be a therapeutic strategy in heart failure to preserve contractile function when the pressure overload cannot be easily alleviated.
Paper by Michael Mutlak MD:
 Mutlak M, Schlesinger-Laufer M, Haas T, Shofti R, Ballan N, Lewis YE, Zuler M, Zohar Y, Caspi LH, Kehat I. Extracellular signal-regulated kinase (ERK) activation preserves cardiac function in pressure overload induced hypertrophy. Int J Cardiol. 2018 May 24
https://www.ncbi.nlm.nih.gov/pubmed/29857938
Mutlak M, Schlesinger-Laufer M, Haas T, Shofti R, Ballan N, Lewis YE, Zuler M, Zohar Y, Caspi LH, Kehat I. Extracellular signal-regulated kinase (ERK) activation preserves cardiac function in pressure overload induced hypertrophy. Int J Cardiol. 2018 May 24
https://www.ncbi.nlm.nih.gov/pubmed/29857938
Genome organization and gene expression in cardiac remodeling
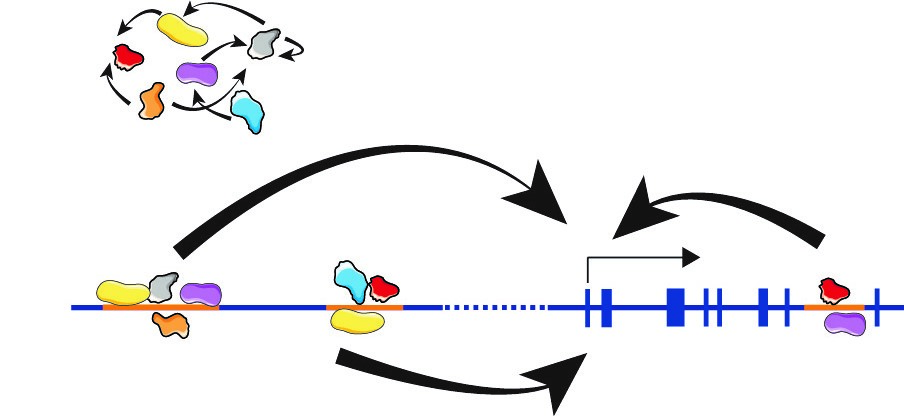 Transcriptional regulation in mammals was simplistically viewed as a process in which soluble protein factors are recruited to promoters, located immediately 5′ of a protein encoding gene, to activate or repress transcription. However, more recent data suggest that the nucleus is an ordered three-dimensional organelle, in which the organization of the genome appears to have implications for orchestrated gene expression. Data suggest that the expression of some genes correlates with their genomic environment. This environment consists of interactions of DNA loci with nuclear structural components such as the nuclear lamina and the nucleoporins and interaction of DNA loci with remote sequences on the same or other chromosomes.
Transcriptional regulation in mammals was simplistically viewed as a process in which soluble protein factors are recruited to promoters, located immediately 5′ of a protein encoding gene, to activate or repress transcription. However, more recent data suggest that the nucleus is an ordered three-dimensional organelle, in which the organization of the genome appears to have implications for orchestrated gene expression. Data suggest that the expression of some genes correlates with their genomic environment. This environment consists of interactions of DNA loci with nuclear structural components such as the nuclear lamina and the nucleoporins and interaction of DNA loci with remote sequences on the same or other chromosomes.
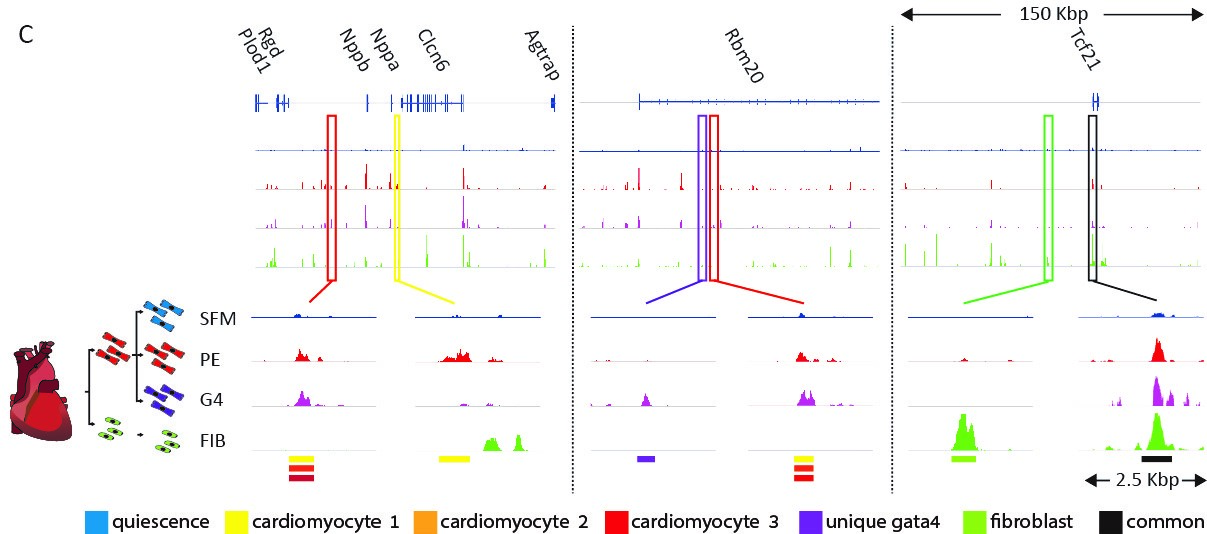 We are trying to understand how cell-type-specific gene expression is controlled by the genome and chromatin in the heart. We performed a genome-wide analysis of regulatory elements (enhancers) in cardiomyocytes and in cardiac fibroblasts using ATAC-seq, H3K27ac ChIP-seq, RNA-seq, and computational transcription factor binding analysis. These analyses enabled us to map the cell-type-specific active enhancers in cardiac fibroblasts and cardiomyocytes and outline the transcription factor families that control them. Our studies showed that in both cell types, distal enhancers, containing concentrated combinatorial clusters of multiple tissue expressed transcription factor recognition motifs, are combinatorically clustered around tissue specific genes.
We are trying to understand how cell-type-specific gene expression is controlled by the genome and chromatin in the heart. We performed a genome-wide analysis of regulatory elements (enhancers) in cardiomyocytes and in cardiac fibroblasts using ATAC-seq, H3K27ac ChIP-seq, RNA-seq, and computational transcription factor binding analysis. These analyses enabled us to map the cell-type-specific active enhancers in cardiac fibroblasts and cardiomyocytes and outline the transcription factor families that control them. Our studies showed that in both cell types, distal enhancers, containing concentrated combinatorial clusters of multiple tissue expressed transcription factor recognition motifs, are combinatorically clustered around tissue specific genes.
Paper be Tal Golan-Lagziel (MD/PhD candidate):

Golan-Lagziel T, Lewis YE, Shkedi O, Douvdevany G, Caspi LH, Kehat I. Analysis of rat cardiac myocytes and fibroblasts identifies combinatorial enhancer organization and transcription factor families. J Mol Cell Cardiol. 2018 Mar;116:91-105.
https://www.ncbi.nlm.nih.gov/pubmed/?term=kehat+and+golan-lagziel https://www.sciencedirect.com/science/article/pii/S0022282818300373?via%3DihubVascular and valve calcification
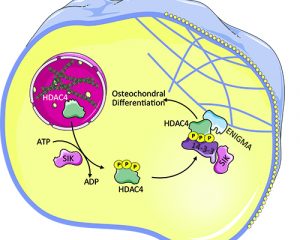 Most aging individuals have progressively enlarging accumulation of calcium in their major arteries. These vascular calcifications, that appear in the walls of blood vessels such as the aorta, coronary, carotid and femoral arteries and in the leaflets of heart valves cause stiffness and impair hemodynamics resulting in hypertension and organ ischemia, valve stenosis, cardiac hypertrophy, and congestive heart failure. The presence of such calcification in any arterial wall is associated with a 3–4-fold higher risk for mortality and cardiovascular events. We are trying to elucidate the molecular mechanisms responsible for vascular and valve calcifications and find novel approaches to stop this devastating process.
We showed that HDAC4 is a positive regulator driving this pathology. HDAC4 can shuttle between the nucleus and cytoplasm, but the cytoplasmic rather than the nuclear activity of HDAC4 promotes calcification. The cytoplasmic location and function of HDAC4 is controlled by the activity of salt-inducible kinase (SIK). In the cytoplasm, HDAC4 binds and its activity depends on the adaptor protein ENIGMA (Pdlim7) to promote vascular calcification. These results establish a cytoplasmic role for HDAC4 and identify HDAC4, SIK, and ENIGMA as mediators of vascular calcification.
Most aging individuals have progressively enlarging accumulation of calcium in their major arteries. These vascular calcifications, that appear in the walls of blood vessels such as the aorta, coronary, carotid and femoral arteries and in the leaflets of heart valves cause stiffness and impair hemodynamics resulting in hypertension and organ ischemia, valve stenosis, cardiac hypertrophy, and congestive heart failure. The presence of such calcification in any arterial wall is associated with a 3–4-fold higher risk for mortality and cardiovascular events. We are trying to elucidate the molecular mechanisms responsible for vascular and valve calcifications and find novel approaches to stop this devastating process.
We showed that HDAC4 is a positive regulator driving this pathology. HDAC4 can shuttle between the nucleus and cytoplasm, but the cytoplasmic rather than the nuclear activity of HDAC4 promotes calcification. The cytoplasmic location and function of HDAC4 is controlled by the activity of salt-inducible kinase (SIK). In the cytoplasm, HDAC4 binds and its activity depends on the adaptor protein ENIGMA (Pdlim7) to promote vascular calcification. These results establish a cytoplasmic role for HDAC4 and identify HDAC4, SIK, and ENIGMA as mediators of vascular calcification.
Paper by Alon Abend (MD/PhD candidate):
 Abend A, Shkedi O, Fertouk M, Caspi LH, Kehat I. Salt-inducible kinase induces cytoplasmic histone deacetylase 4 to promote vascular calcification. EMBO Rep. 2017 Jul;18(7):1166-1185
https://www.ncbi.nlm.nih.gov/pubmed/28588072
http://embor.embopress.org/content/18/7/1166.long
Abend A, Shkedi O, Fertouk M, Caspi LH, Kehat I. Salt-inducible kinase induces cytoplasmic histone deacetylase 4 to promote vascular calcification. EMBO Rep. 2017 Jul;18(7):1166-1185
https://www.ncbi.nlm.nih.gov/pubmed/28588072
http://embor.embopress.org/content/18/7/1166.long